PREPRINT VERSION AT Journal of Animal Science and Biotechnology
Yentel Mateo-Otero, Pol Fernández-López, Ariadna Delgado-Bermúdez, Pau Nolis, Jordi Roca, Jordi Miró, Isabel Barranco, Marc Yeste
Background
Metabolomic approaches, which include the study of low molecular weight molecules, is an emerging -omics technology useful for the identification of biomarkers. In this field, nuclear magnetic resonance (NMR) spectroscopy approach has already been used to uncover (in)fertility biomarkers in the seminal plasma (SP) of several mammalian species. However, NMR studies profiling SP metabolome to uncover in vivo fertility biomarkers are yet to be carried out in pigs. Thus, this study aimed to evaluate the putative relationship between the presence/concentration of SP-metabolites and in vivo fertility outcomes. To this end, 24 entire ejaculates (three ejaculates per boar) were collected from artificial insemination (AI)-boars throughout a year (one ejaculate every four months). Immediately after collection, ejaculates were centrifuged (1,500×g for 10 min twice) to obtain SP-samples and were stored (− 80°C) for subsequent metabolomic analysis by NMR spectroscopy. Fertility outcomes from 1,525 inseminations were recorded over a year, including farrowing rate, litter size, stillbirths per litter and the duration of pregnancy. These data were corrected to isolate the direct boar effect on each in vivo fertility parameter using a multivariate statistical model.
Results
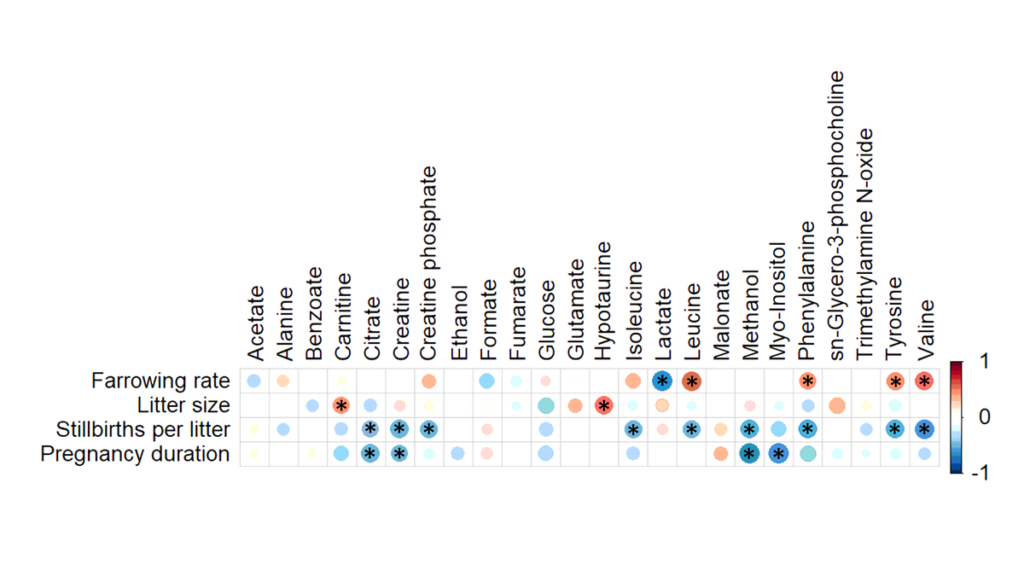
A total of 24 metabolites were identified and quantified in all SP-samples. ROC curve analysis showed that lactate levels in SP had discriminative capacity for farrowing rate (area under the curve (AUC) = 0.764; P < 0.05) while carnitine (AUC = 0.847), hypotaurine (AUC = 0.819), sn-glycero-3-phosphocholine (AUC = 0.833), glutamate (AUC = 0.799) and glucose (AUC = 0.750) had it for litter size (P < 0.05). Similarly, citrate (AUC = 0.743), creatine (AUC = 0.812), phenylalanine (AUC = 0.750), tyrosine (AUC = 0.753) and malonate (AUC = 0.868) levels had discriminative capacity for stillbirths per litter (P < 0.05); and malonate (AUC = 0.767) and fumarate (AUC = 0.868) concentrations for gestation length (P < 0.05).
Conclusions
Considering these results, the assessment of selected SP-metabolites in ejaculates through NMR spectroscopy could be considered as a promising non-invasive tool to predict in vivo fertility outcomes in pigs. Moreover, supplementing AI-doses with specific metabolites should also be contemplated as a way to improve their fertility potential.