Mononuclear ruthenium compounds bearing N-donor and N-heterocyclic carbene ligands: structure and oxidative catalysis
Mononuclear ruthenium compounds bearing N-donor and N-heterocyclic carbene ligands: structure and oxidative catalysis
DOI: 10.1039/C6DT04729G
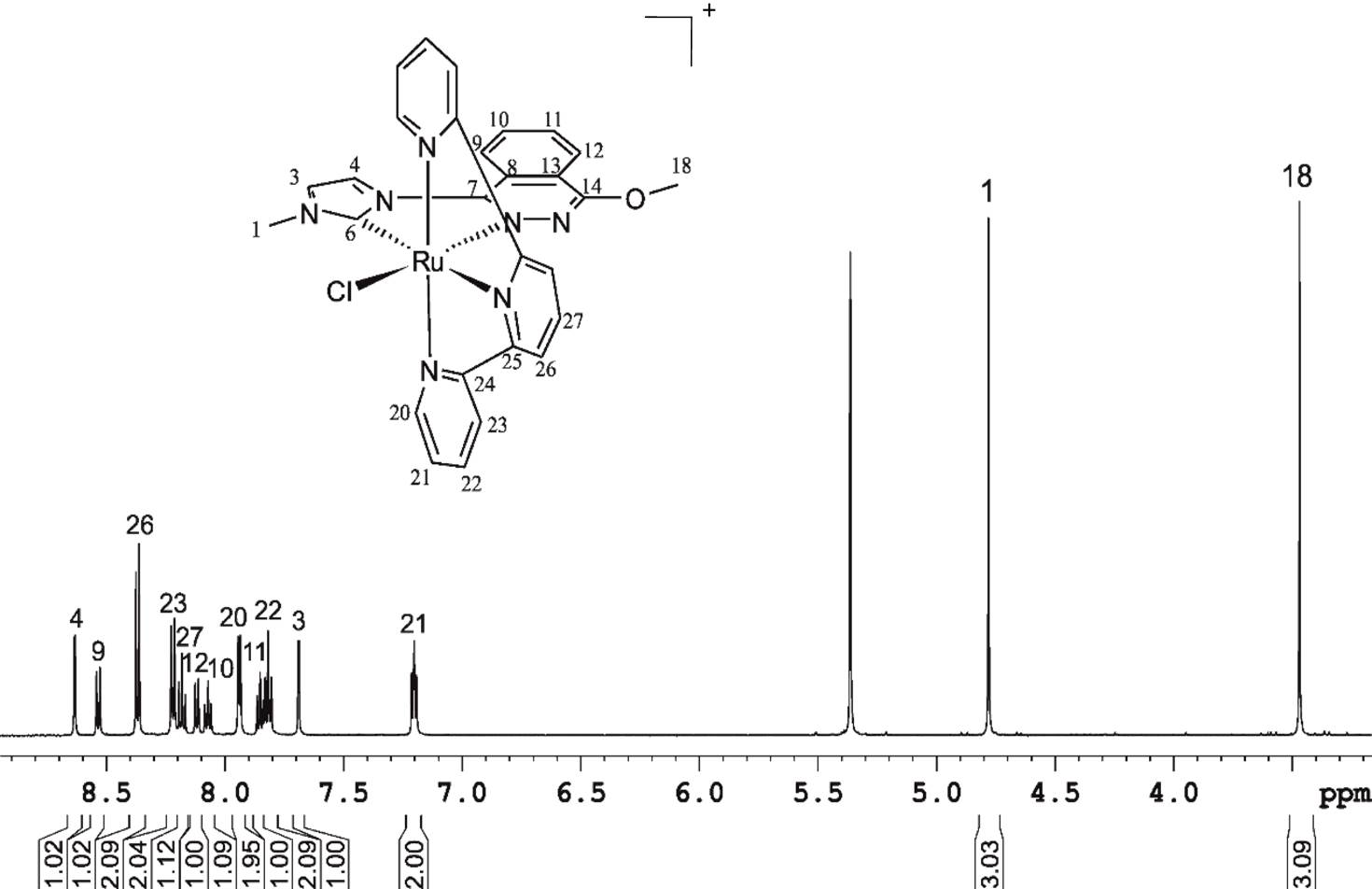
ABSTRACT A new CNNC carbene-phthalazine tetradentate ligand has been synthesised, which under reaction with [Ru(T)Cl3] (T = trpy, tpm, bpea; trpy = 2,2′;6′,2″-terpyridine; tpm = tris(pyrazol-1-yl)methane; bpea = N,N-bis(pyridin-2-ylmethyl)ethanamine) in MeOH or iPrOH undergoes a C-N bond scission due to the nucleophilic attack of a solvent molecule, with the subsequent formation of the mononuclear complexes cis-[Ru(PhthaPz-OR)(trpy)X]n+, [Ru(PhthaPz-OMe)(tpm)X]n+ and trans,fac-[Ru(PhthaPz-OMe)(bpea)X]n+ (X = Cl, n = 1; X = H2O, n = 2; PhthaPz-OR = 1-(4-alkoxyphthalazin-1-yl)-3-methyl-1H-imidazol-3-ium), named 1a+/2a2+ (R = Me), 1b+/2b2+ (R = iPr), 3+/42+ and 5+/62+, respectively. Interestingly, regulation of the stability regions of the different Ru oxidation states is obtained by the different ligand combinations, going from 62+, where Ru(III) is clearly stable and mono-electronic transfers are favoured, to 2a2+/2b2+, where Ru(III) is almost unstable with regards to its disproportion. The catalytic performance of the Ru-OH2 complexes in chemical water oxidation at pH 1.0 points to poor stability (ligand oxidation), with subsequent evolution of CO2 together with O2, especially for 42+ and 62+. In electrochemically driven water oxidation, the highest TOF values are obtained for 2a2+ at pH 1.0. In alkene epoxidation, complexes favouring bi-electronic transfer processes show better performances and selectivities than those favouring mono-electronic transfers, while alkenes containing electron-donor groups promote better performances than those bearing electron-withdrawers. Finally, when cis-β-methylstyrene is employed as substrate, no cis/trans isomerization takes place, thus indicating the existence of a stereospecific process.